- RESEARCH
- Open access
- Published:
Establishment of a yeast-based VLP platform for antigen presentation
Microbial Cell Factories volume 17, Article number: 17 (2018)
Abstract
Background
Chimeric virus-like particles (VLP) allow the display of foreign antigens on their surface and have proved valuable in the development of safe subunit vaccines or drug delivery. However, finding an inexpensive production system and a VLP scaffold that allows stable incorporation of diverse, large foreign antigens are major challenges in this field.
Results
In this study, a versatile and cost-effective platform for chimeric VLP development was established. The membrane integral small surface protein (dS) of the duck hepatitis B virus was chosen as VLP scaffold and the industrially applied and safe yeast Hansenula polymorpha (syn. Pichia angusta, Ogataea polymorpha) as the heterologous expression host. Eight different, large molecular weight antigens of up to 412 amino acids derived from four animal-infecting viruses were genetically fused to the dS and recombinant production strains were isolated. In all cases, the fusion protein was well expressed and upon co-production with dS, chimeric VLP containing both proteins could be generated. Purification was accomplished by a downstream process adapted from the production of a recombinant hepatitis B VLP vaccine. Chimeric VLP were up to 95% pure on protein level and contained up to 33% fusion protein. Immunological data supported surface exposure of the foreign antigens on the native VLP. Approximately 40 mg of chimeric VLP per 100 g dry cell weight could be isolated. This is highly comparable to values reported for the optimized production of human hepatitis B VLP. Purified chimeric VLP were shown to be essentially stable for 6 months at 4 °C.
Conclusions
The dS-based VLP scaffold tolerates the incorporation of a variety of large molecular weight foreign protein sequences. It is applicable for the display of highly immunogenic antigens originating from a variety of pathogens. The yeast-based production system allows cost-effective production that is not limited to small-scale fundamental research. Thus, the dS-based VLP platform is highly efficient for antigen presentation and should be considered in the development of future vaccines.
Background
Since the 1980s virus-like particles (VLP) have been known for their immunogenic properties [1] and have been well established as safe, effective vaccines and drug delivery systems in humans [2,3,4]. VLP induce strong humoral immune and T cell responses but they lack the risks of conventional vaccines: they do not contain genetic material and are unable to replicate [5,6,7]. Recombinant VLP are highly valued as vaccine development platforms and VLP scaffolds are used to display immunogenic antigens originating from foreign pathogens (referred to as chimeric VLP) [8].
Vaccination plays a leading role in preventing infectious diseases in animals and improving animal welfare [9]. Aside from economic advantages, vaccination allows the reduced use of antibiotics in animal farming and thus helps to prevent the spread of antibiotic resistances in the environment [10]. In the veterinary sector, conventional vaccines are still predominant. Subunit vaccines based on soluble, monomeric proteins often have limitations regarding immunogenicity which can be optimized by VLP-based approaches [11, 12].
Our current study describes the establishment of a novel and versatile Hansenula-based VLP platform. We chose the membrane integral small surface protein (dS) of the duck hepatitis B virus (DHBV) as scaffold protein for chimeric VLP production [13, 14]. It allows the development and high-yield production of chimeric VLP which tolerate the incorporation of a variety of large foreign antigens. Thus, key challenges for VLP-based vaccine development are addressed and fulfilled using this platform, which has not been reported for other VLP platforms before [6, 15, 16].
The DHBV is closely related to the human hepatitis B virus (HBV) and the virions are of comparable size (42–50 nm in diameter) and structure [17]. However, size and composition of their subviral particles are differing which certainly induces differences in their recombinant counterparts, too. The naturally occurring VLP from the DHBV are described as 35–60 nm particles [18] and the ratio of the large to the small DHBV surface proteins within the VLP is identical (approximately 1:4, [19, 20]) to that found in the virions’ envelope [21]. In contrast, the spherical HBV VLP are smaller (~ 22 nm diameter) and the small surface protein (HBsAg) is enriched compared to the composition of the virions’ envelope [21]. Additionally, dS VLP are lacking an equivalent antigen to the highly immunogenic “a determinant” of the HBsAg that predominates the host’s immune reaction [20, 22, 23].
As a microbial cell factory, we chose the methylotrophic yeast Hansenula polymorpha (H. polymorpha, syn. Pichia angusta, Ogataea polymorpha, [24]). In the field of single-layer VLP production, advantages of yeast-based systems over mammalian [25, 26], bacterial and baculovirus/insect cell systems [27, 28] are widely known. In particular, H. polymorpha is established as safe microbial cell factory for recombinant products that have been granted “generally recognized as safe” (GRAS) status and for the production of biopharmaceuticals like hepatitis B VLP vaccines [29,30,31].
Another focus of this project was the development of a VLP platform suited for the production of VLP-based vaccines suitable for the application in the veterinary sector. Hence, it is compatible with the “differentiating infected from vaccinated animals” (DIVA) strategy and independent of antibiotic resistance genes during all stages of development and production [32].
Firstly, plain dS VLP (without a foreign antigen displayed) were purified at several mg scale as a proof-of-principle and benchmark. For this purpose, a downstream process (DSP) approved for hepatitis B vaccine production from yeast [31] was applied. Analytical tools for specific detection of dS, quantification of host cell protein (HCP) impurities and VLP characterization were established in parallel. Antigens of up to 412 amino acids (aa) were chosen to be incorporated in the dS VLP scaffold to test the versatility of the platform for chimeric VLP production. The chosen antigens originated from the following four different viruses that cause infectious diseases in animals:
-
1.
The bovine viral diarrhea virus (BVDV) is an important pathogen of cattle, also infecting sheep and pigs. It is responsible for significant animal suffering and economic losses worldwide [33].
-
2.
The classical swine fever virus (CSFV) is acknowledged as a global threat for swine [34] and is listed as notifiable animal diseases by the World Organization for Animal Health.
-
3.
The feline leukemia virus (FeLV) is a retrovirus threatening domestic cats [35].
-
4.
The west nile virus (WNV) is a mosquito vector transmitted zoonotic virus of the Flaviviridae family. It circulates in birds as natural hosts but can be transmitted to mammals including humans causing west nile fever [36]. WNV could represent a case example because of its close relationship to the yellow fever and dengue virus, which cause two of the most important mosquito-borne human diseases [37].
Antigen-presenting chimeric VLP were rationally engineered by genetic fusion of foreign antigens to either the C- or N-terminus of the dS. Co-expression of the fusion proteins with the VLP-forming scaffold protein allowed the isolation of chimeric VLP in all cases. Compared to other chimeric VLP platforms, no linker [38] or chemical coupling of the antigen to the VLP scaffold [39] was required. Thus, the use of the dS allowed us to minimize the complexity of the chimeric VLP to the essentials.
The methodology applied for purification of plain dS VLP could widely be transferred to chimeric VLP displaying the different foreign antigens. A variety of analyses regarding particle structure and stability were performed for different VLP preparations. For chimeric VLP, a shelf life of at least 6 months and resistance to temperature-induced stress comparable to that of plain dS VLP were demonstrated.
Methods
Genes, vectors, cloning
The designed open reading frames (ORF) encoding the dS (Genbank accession number: MF510122) and the different fusion proteins were synthesized by GeneArt/Life Technologies (Regensburg, Germany). They were flanked with EcoRI and BamHI restriction sites and codon-optimized for recombinant expression in H. polymorpha. Genbank accession numbers of donor sequences are given in Table 2. Synthesized ORF were inserted between the EcoRI and BamHI sites of the antibiotic resistance marker free H. polymorpha expression plasmid pB14 [40] or a derivative thereof with LEU2 instead URA3 gene for selection in yeast. Cloning was done in an E. coli K12 derivative (genotype: F-pyrF74:Tn5 supE44 lacY1 ara-14 galK2 xyl-5 mtl-l leuB6 proA2 hsdS20 recA13 rpsL20 thi-1 lambda-) purchased from DSMZ (No. DSM 6201, Braunschweig, Germany). It is optimized for cloning of yeast shuttle vectors containing LEU2 and/or URA3. Chemically competent bacteria were transformed by a heat shock protocol (60 s, 40 °C [41]). For plasmid amplification, strains were grown at 37 °C in M9-based minimal medium [42] supplemented with amino acids (mg L−1). l-Arginine (10), l-histidine (5), l-isoleucine (30), l-leucine (30), l-methionine (5), l-proline (20), l-threonine (25), l-tryptophan (20), d/l-phenylalanine (30), l-lysine-monohydrate (20) and l-leucine (30).
Heterologous yeast strain generation
The auxotrophic H. polymorpha strains ALU3 (relevant genotype: ade1, leu2, ura3) [43] and RB11 (relevant genotype: ura3) [43] were used as expression hosts. They are derivatives of wild type strain ATCC® 34438™ (CBS 4732, IFO 1476, JCM 3621, NBRC 1476, NCYC 1457, NRRL Y-5445) [44]. Yeast transformation was performed by electroporation [45] and subsequent strain generation and isolation [46]. Thereby, the expression plasmids integrated genomically stable in different copy numbers into the host genome. Heterologous yeast strains were stored as glycerol stocks at − 80 °C.
Expression studies and VLP diagnosis
Screening for heterologous H. polymorpha production strains was performed at 37 °C in 3 mL test tube scale. Pre-cultures were grown in YPD medium to stationary phase and used to inoculate YPG medium containing 20 g L−1 glycerol (AppliChem, Darmstadt, Germany) as carbon source. After a derepression phase of 56 h, 1% (v/v) methanol (AppliChem, Darmstadt, Germany) was added and cultivation was extended for additional 24 h. Cells were harvested by centrifugation (6000g, 15 min, 4 °C) and disrupted by glass beads (0.5–0.7 mm, Willy A. Bachofen, Nidderau-Heldenberg, Germany) in 1.5 mL reaction tubes on a shaker (basic Vibrax® shaker, IKA®-Werke, Staufen, Germany) at maximal frequency for 30 min at 4 °C.
To analyze whether the fusion proteins and the dS co-expressed in H. polymorpha are involved in chimeric VLP formation, two subsequent ultracentrifugation steps were accomplished in Optima™ L90K centrifuge (rotor type: 70.1 Ti, tubes: 16 * 76 mm, Beckman Coulter, Brea, California, USA). After cell disruption, the soluble protein fractions were prepared and layered on top of a sucrose cushions (2 mL 70% (w/v); 3 mL 20% (w/v), [47]). The boundary layers between the two sucrose layers were harvested after ultracentrifugation (90 min, 51,000 rpm, 18 °C). These fractions were subsequently mixed with 6 M CsCl (AppliChem, Darmstadt, Germany) stock solution to 1.5 M final CsCl concentration. Mixtures were subjected to density gradient separation (65 h at 48,400 rpm, 4 °C). Thereafter, 11 fractions were collected according to their densities and analyzed by Western blot to specifically identify the product containing fractions. As indication for chimeric VLP formation were regarded: (1) accumulation of the product proteins in the boundary layer of the sucrose cushion ultracentrifugation. (2) Co-separation of the dS and the respective fusion protein from contaminating HCP. (3) Gravimetrically determined densities of 1.1–1.2 g cm−3 of the product containing fractions.
Heterologous protein production and purification of VLP at laboratory scale
Cell mass used for pilot VLP production process was generated in a 2.5 L scale fed-batch fermentation using a stirred tank (Labfors 5, Infors, Bottmingen, Switzerland). The bioreactor was sterilized by autoclaving after filling with 2.5 L animal component free complex medium containing 20 g L−1 yeast extract (BD Biosciences, Heidelberg, Germany), 40 g L−1 peptone from soymeal (AppliChem, Darmstadt Germany), 20 g L−1 glycerol, 3.4 g L−1 yeast nitrogen base (YNB, Becton, Dickinson Difco™, Franklin Lakes, USA), 10 g L−1 ammonium sulfate (AppliChem, Darmstadt Germany), 0.5 g L−1 adenine and 2 g L−1 leucine. Aqueous solutions of NH3 (12.5% (w/w), sterile filtered) and H3PO4 (28% (w/w), Merck, Darmstadt, Germany) were used as corrective media to keep pH constant (set point 6.2) throughout fermentation. Struktol J 673 (10% (v/v) aqueous solution, Schill + Seilacher, Hamburg, Germany) was utilized as antifoam agent. Temperature was kept constant at 37 °C, aeration was adjusted to 1 vvm (2.5 NL min−1) and the pO2 setpoint was 40%. The medium was inoculated using shake flask pre-cultures. After a batch phase of 12 h, 360 mL of 750 g L−1 glycerol solution were fed continuously over 37 h. Target gene expression was induced by pulse-wise addition of 65 mL 28.5% (w/w) glycerol and 71.5% (w/w) methanol solution. Cells were harvested after 70.5 h total fermentation time by centrifugation (30 min, 4 °C, 17,000g), washed with wash buffer (50 mM Na-phosphate buffer, 2 mM EDTA, pH 8.0) and stored at − 20 °C until further processing.
The dry cell weight (dcw) was quantified using a moisture analyzer (MLS 50-3 HA250, Kern & Sohn, Balingen, Germany). OD600 of cell suspensions was determined with a spectrophotometer (DU 640 Beckman Coulter, Brea, California, USA).
Plain dS VLP and chimeric VLP with the dS as scaffold were purified by a DSP invented for purification of HBsAg VLP [31] including adjustments due to down-scaling of the process to laboratory scale. Briefly, PEG6000 and NaCl (AppliChem, Darmstadt, Germany) were added to crude cell lysate after yeast cell disruption by six cycles of high pressure homogenization (~ 1500 bar, APV 2000, SPX Flow Technology, Unna, Germany) in presence of 2 mM PMSF. The mixture was incubated over-night at 4 °C and then centrifuged (17,000g, 30 min, 4 °C). Subsequently, 15 g L−1 fumed silica matrix Aerosil (type 380 V, Evonik, Essen, Germany) was added to the soluble protein fraction (PEG-SN). Product adsorption to Aerosil was allowed over-night at 4 °C during incubation on magnetic stirrer MR3001 (Heidolph Instruments, Schwabach, Germany). The matrix was washed with 77 mM NaCl aqueous solution volume-normalized to the PEG-SN. A buffer for desorption of the product from the Aerosil was added (10 mM di-sodium tetraborate decahydrate, 2 mM EDTA, 6 mM deoxycholic acid sodium salt, pH 9.1) using a quarter of the PEG-SN volume. The suspension was stirred for 1 h at 55 °C. Only in the case of plain dS VLP, the soluble product fraction (desorbate) was applied to anion exchange chromatography (Mustang Q XT, PALL Life Sciences, Port Washington, New York, United States) and eluted with 0.5 M NaCl. Product containing fractions were pooled and concentrated by ultrafiltration (Vivaspin® sample concentrator, MWCO 100 kDa, Sigma-Aldrich, Steinheim am Albuch, Germany) and applied to CsCl density gradient separation as a final purification step. Product containing fractions were pooled, desalted by dialysis (Slyde-A-Lyzer™ dialysis cassettes, MWCO 20 kDa, Thermo Fisher Scientific, Waltham, USA) against desalting buffer (8 mM Na-phosphate buffer pH 7, 154 mM NaCl, AppliChem, Darmstadt, Germany) and sterile filtered (Filtropur S 0.2 filters, Sarstedt, Nümbrecht, Germany).
For chimeric VLP preparations, the desorbate was concentrated by ultrafiltration (Minimate™ TFF tangential flow filtration Capsule Omega 100 k Membrane, PALL, Port Washington, New York, United States) and directly applied to CsCl density gradient separation.
Particle characterization
Particle size distribution of VLP preparations was analyzed by dynamic light scattering (DLS) using a DelsaMax CORE (BCI-3161-DMC) system operating at 25 °C and equipped with a 100 mW 658 nm diode laser along with disposable cuvettes (Beckman Coulter, Brea, California, USA). Presented data are mean values from 10 acquisitions. Stability assessments at elevated temperatures were completed by step-wise increase of chamber temperature by 5 °C. Before collecting data as described before, temperature equilibration was allowed for of 5 min. Increase of temperature was continued until aggregation was detected.
Transmission electron microscopy (TEM) was used for analysis of the shape and integrity of the VLP. Volumes of 15 µL fixative (4% paraformaldehyde, 0.1 M cacodylate buffer, pH 7.2) were mixed with 15 µL of purified VLP samples. Mixtures were incubated for 15 min at room temperature (RT). Then, 3 µL of the mix were transferred to a nickel grid coated with Formvar and carbon. After 2 min of incubation at RT, the remaining liquid was removed carefully with absorbent paper and the grid was washed twice with 30 µL of distilled water and equilibrated with 30 µL staining solution (1.5% (w/v) uranyl acetate aqueous solution). The liquid was immediately removed, and the samples were stained by incubating the grids for 30 s with 30 µL staining solution. After drying at RT for at least 30 min, TEM images were generated with H600 TEM (Hitachi, Tokyo, Japan) at 75 kV.
Super-resolution microscopy (structured-illumination microscopy; N-SIM) was used to investigate co-localization and surface exposure of the scaffold protein dS and the foreign antigen in nano-scale structures. Chambered slides (Nunc) were coated with 0.01% poly-l-lysine (Sigma-Aldrich, Steinheim am Albuch, Germany) for 20 min before washing thrice with PBS. Native VLP samples were added to the coated wells. They were allowed to settle over-night at 4 °C. The supernatant of unbound VLP were removed and samples were fixed with 4% paraformaldehyde for 20 min before washing thrice with PBS. Samples were blocked with 6% bovine serum albumin (Sigma-Aldrich, Steinheim am Albuch, Germany) for 20 min and washed thrice with PBS. Samples were dual-labeled with primary antibodies biotinylated anti-dS mAb (7C12) and anti-CSFV E2 mAb (PrioMab CSFV V8 Monoclonal Antibody, Thermo Fisher Scientific, Waltham, USA) and subsequently, secondary labeled with streptavidin-488 (Invitrogen, Carlsbad, California, USA, green fluorescence) and anti-mouse AlexaFluor 594 (red fluorescence, Invitrogen, Carlsbad, California, USA). Samples were subjected to another fixation step with 4% paraformaldehyde for 10 min. The super-resolution images were collected using a Nikon N-SIM microscope equipped with 488, 561 and 640 nm lasers, an Andor iXON DU897 EM-CCD camera and a oil immersion lens (100-fold magnification) having a numerical aperture of 1.49. The z-series was acquired using NIS-Elements and analysed both using NIS-Elements and the open java source, ImageJ/FIJI.
Quantification of proteins and lipids
Protein concentrations were determined by precipitation Lowry protein assay [48]. Samples were analyzed at least as triplicates. Commercial BSA stock solution (Sigma-Aldrich, Steinheim am Albuch, Germany) was used as standard. Lipid content of VLP preparations was determined based on sulfo-phospho-vanillin reaction [49] with refined soya oil (Caesar & Loretz GmbH, Hilden, Germany) used as standard.
SDS-PAGE, Western blot and dot blot analysis
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate proteins according to their size. The Criterion™ Cell system was used (BioRad, München, Germany). Consumables were Criterion™ XT precast gels (4–12% Bis–Tris), XT reducing agents, XT sample buffer and Precision Plus Protein™ Prestained Standard All Blue as molecular weight (MW) standard. Polyacrylamide (PAA) gels were either stained with Coomassie dye [50] or subjected to Western blot analysis for specific detection of VLP proteins. The proteins were transferred onto cellulose nitrate membranes (Sartorius Stedim Biotech, Göttingen, Germany) by semi dry blotting [51]. Prior to immunostaining, the membrane was incubated with Ponceau S solution (AppliChem, Darmstadt Germany) for reversible, non-specific staining of the transferred proteins. After de-staining in PBS and blocking of the membranes, the monoclonal antibodies listed in Table 1 were used as primary antibodies. The detection system was completed with appropriate secondary antibodies (BioRad, München, Germany) conjugated with alkaline phosphatase along with BCIP-NBT solution (VWR international, Radnor, USA). The reactivity of native VLP samples was assessed in dot blot assays without any denaturing treatment and applied to a nitrocellulose membrane by vacuum. The membranes were immune stained as described for Western blot analysis. A commercial software package was used for analysis by densitometry (Image Lab™, BioRad, München, Germany).
Analysis of HCP
Host cell protein (HCP) content of VLP preparations was analyzed by anti-HCP Western blot and enzyme-linked immunosorbent assay (ELISA). A polyclonal antiserum isolated from goats immunized with H. polymorpha HCP (Artes Biotechnology, Langenfeld, Germany/BioGenes, Berlin, Germany) was used in both cases as primary immunoreagent. The detection system for Western blot analysis was completed with a rabbit anti-goat IgG AP conjugate (BioRad, München, Germany) in combination with BCIP-NBT solution.
HCP quantification was done by an indirect ELISA in high binding plates (Sarstedt, Nümbrecht, Germany). Crude cell extract of a H. polymorpha vector control strain was used for calibration. The ELISA plate was first coated with the samples under investigation and then immunodecorated with the anti-HCP serum. Subsequently an enhanced streapavidin/biotin system was employed: the ELISA plates were incubated with biotinylated anti-goat polyclonal antibodies raised in rabbits (KPL, Milford, Massachusetts, USA) as secondary antibody. Then, streptavidin-HRP (GE Healthcare, Amersham, UK) was added and ABTS substrate solution was used for colorization (BioRad, München, Germany).
Protein deglycosylation assay
N-Glycosylation of the heterologous target proteins was analyzed by treatment with an endoglycosidase H (EndoH) prior to SDS-PAGE and Western blotting. Protein samples were denatured (95 °C for 5 min) in glycoprotein denaturing buffer (New England Biolabs, Frankfurt a. M., Germany) and subsequently treated with EndoH in glyco3 buffer (New England Biolabs, Frankfurt a. M., Germany) at 37 °C for 60 min. A shift of target protein-specific signals in Western blot analysis to lower apparent MW compared to the untreated sample indicated N-glycosylation of the target protein.
Results
Design of fusion proteins for chimeric VLP production
Typically, co-production of the VLP scaffold protein dS and fusion proteins composed of a foreign antigen C- or N-terminally fused to the dS is required for chimeric VLP production. To demonstrate it with an application-oriented focus, a variety of fusion proteins were designed harboring immunogenic antigens originated from the animal infecting viruses BVDV, CSFV, FeLV or WNV (Table 2).
The envelope protein E2 appeared as valuable antigen for targeting BVDV and CSFV. For both viruses, E2 was described to be the key immunogen involved in neutralization upon infection [52,53,54,55]. To vary the complexity of the constructed fusion proteins, N-terminal parts of the respective E2 of different lengths were chosen to be displayed on the VLPs’ surface. For the fusion protein design, information on structures and immunogenic domains was considered [56,57,58]. In the longer fusion protein variants which contained N-glycosylation motifs, the leader peptide of the chicken lysozyme (CL) was included. Thus, fusion proteins were targeted to the secretory machinery of the yeast [59] which enhanced protein N-glycosylation of the constructs to improve their immunogenic potential [60].
The protein p45, especially in combination with p15E, was reported to protect cats from FeLV infection [61,62,63]. Both antigens were included in our project as well as domain III of the WNV envelope protein E which is also known as potent immunogen [64].
Co-expression of the dS and the designed fusion proteins
Aiming at co-expression of the dS and the fusion proteins for recombinant chimeric VLP production, the following three strain generation strategies were applied. The constructed expression plasmids were based on pB14 which is free of antibiotic resistance genes [40].
-
I.
Staggered transformation: firstly, a dS-encoding expression plasmid was introduced into ALU3 and a strain producing dS was isolated and cryo-preserved at − 80 °C. In a second transformation, an expression plasmid encoding the fusion protein of choice was introduced into the selected dS-producing strain. Strains co-producing dS and the fusion protein could then be isolated.
-
II.
Co-transformation: ALU3 was transformed in one electro-transformation with two pB14-based plasmids or plasmid fragments, respectively. One of them encoded the dS the other encoded the respective fusion protein. The plasmids or plasmid fragments carried unequal selection markers.
-
III.
Dual plasmid approach: transformation of ALU3 or RB11, respectively, with the novel pB14-2xFPMT-dS expression plasmid (Fig. 1) encoding both, the dS and a fusion protein after insertion of an appropriate gene.
Fig. 1 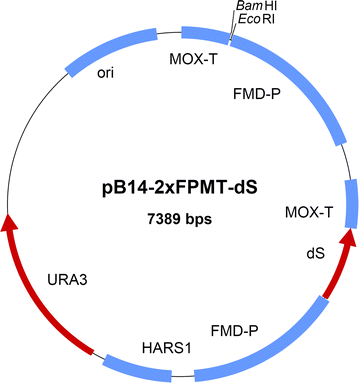
Map of the novel expression plasmid pB14-2xFPMT-dS carrying two expression cassettes specifically tailored to heterologous co-production of the dS and a fusion protein for chimeric VLP production with H. polymorpha. The dS encoding gene is stably inserted. The ORF encoding the desired fusion protein is to be inserted using EcoRI and BamHI sites. The S. cerevisiae URA3 gene was used for selection of bacteria and yeast. Further features: ori, origin of replication; HARS1, H. polymorpha autonomously replicating sequence 1; FMD-P, FMD promoter; MOX, MOX terminator both derived from H. polymorpha genome

All three strain generation strategies yielded strains co-producing the dS and each of the designed fusion proteins (Table 2). The dS and the dS domain of the fusion protein respectively, were specifically detected by anti-dS Western blots. This is exemplarily shown for co-production of dS and E2CSFV102-dS in Fig. 2. Strains producing both heterologous proteins at approximately equal levels (lane 7) could be identified as well as strains producing one of them in excess (e.g. lanes 1 and 16). The dS expression levels of the strains generated by transformation with pB14-based plasmids without an antibiotic resistance gene reached (e.g. lane 1) or surpassed (e.g. lane 16) the productivity of the reference strain A#299 (lane 2) generated with a pFPMT121-based plasmid encoding the dS. Finally, for each fusion protein one strain has been selected and used for further analyses. Designated strains are listed in Table 4. Transformation of strain M#22-8 with a FeLVp15E-dS encoding expression plasmid yielded strain M#4-5 which co-expresses the three heterologous proteins dS, FeLVp45-dS and FeLVp15E-dS.

Western blot analysis of crude cell lysates probed with anti-dS mAB 7C12. Samples originated from 17 different recombinant H. polymorpha strains co-producing the dS and the fusion protein E2CSFV102-dS (lanes 1, 3–17) or producing only the dS (lane 2, strain A#299). Indicated strain generation strategies (I), (II) and (III) refer to the methods described in the text. Strains A#299 and D#79 (highlighted by arrows) were used for VLP production. M, molecular weight marker
Each of the strain generation strategies (I), (II) and (III) yielded heterologous yeast strains which stably co-produced the dS and a fusion protein within only one experimental sequence of transformation and selection. However, the strategies had their individual advantages summarized in Table 3. The staggered transformation approach (I) reduced lab work to a minimum since only a small state-of-the art expression plasmid encoding the fusion protein needed to be assembled and verified by sequencing. Also, transformation efficiency and frequency of strains co-producing the two desired proteins were maximal. The greatest variety of expression levels of the two target proteins was observed among strains isolated from strategy (II). However, transformation efficiency was lower compared to strategy (I) and (III): two plasmids with two unequal selection markers were used in the same transformation and only transformants having both plasmids incorporated were selected. For strategy (III) only one auxotrophy marker (ura3) was needed. This expanded the choice of applicable host strains to e.g. the industrially established strain RB11. Transformation of strain RB11 with the dual plasmid (Fig. 1) yielded directly prototrophic production strains. Nevertheless, the large plasmid harboring multiple homologous sequences (FMD promoter, MOX terminator, dS encoding sequence, Fig. 1) complicated cloning work and especially sequencing of the ORF encoding the fusion protein.
Solubilization of the target proteins
Interestingly, in case of co-expression of dS and E2CSFV102-dS, the relative expression levels of both target proteins had an impact on their solubilization during cell disruption. If the dS was produced in excess over E2CSFV102-dS more than 80% of both heterologous proteins were found solubilized in the supernatant after cell disruption. However, if the protein levels were equal or if the fusion protein was produced in excess, both product proteins were found mainly insoluble (data not shown). In the DSP for VLP purification adopted from HBsAg VLP vaccine production [31], the supernatant after cell disruption is processed. Due to the improved product solubilization in strains producing the dS in excess over E2CSFV102-dS, strain D#79 (Fig. 2, lane 16) was chosen for production of chimeric VLP in mg scale displaying the CSFV antigen as described below.
Detection of target protein N-glycosylation
Samples derived from each of the strains listed in Table 4 were subjected to the protein deglycosylation assay. Analysis of crude cell extract from strains D#53 and D#73 co-producing the dS and the fusion proteins E2CSFV337-dS or E2CSFV184-dS, respectively, are representatively shown in Fig. 3a, b. In both cases, one dominant dS-specific signal at approximately 14 kDa and therewith slightly below its theoretical MW (18.2 kDa) was accompanied with three weaker signals of higher mobility (dS-HMF). The comparisons of lanes 1a/b and 2a/b demonstrated that the dS-specific signals did not respond to EndoH treatment. This showed that the one potential N-glycosylation site present in the dS amino acid sequence was not occupied. Moreover, the occurrence of the dS-HMF could be influenced by treatment with different detergents indicating that they do not represent truncated species of the dS (data not shown).

Protein deglycosylation assay of crude cell lysates derived from recombinant H. polymorpha strains D#53 (a) and D#73 (b) co-producing the dS and the fusion proteins E2CSFV337-dS or E2CSFV184-dS, respectively. Samples were analyzed by Western blot probed with anti-dS mAB 7C12 with (lanes 1a and 1b) or without (lanes 2a and 2b) previous EndoH treatment. M, molecular weight marker
In contrast, the fusion protein-specific bands were sensitive to treatment with EndoH. In the samples not treated with EndoH, the fusion proteins E2CSFV337-dS or E2CSFV184-dS appeared as clusters of distinct bands. Upon protein deglycosylation by incubation with EndoH (lanes 2a and 2b), the signals merged into one single band corresponding to the fusion protein-specific signal of lowest MW detected in lanes 1a or 1b, respectively. This indicated N-glycans cleaved off the fusion proteins and agrees with the presence of five or two potential N-glycosylation sites in the amino acid sequences. Glycosylation of the CSFV antigens demonstrated that they have been exposed to the lumen of the endoplasmic reticulum (ER) or Golgi system, the compartments of protein N-glycosylation. Analysis of the other designated strains producing the different fusion proteins is summarized in Table 4.
Identity of the foreign antigens
The identities of the foreign antigens were confirmed for all fusion proteins except the FelV-derived p15E-dS and p45-dS and the dS-E2BVDV196 by Western blot analysis applying monoclonal antibodies specific for the foreign antigens. In all cases the fusion proteins but not the unfused dS were detected. This is exemplarily shown in Fig. 4 for the three fusion proteins containing CSFV-derived antigens. For the fusion protein E2CSFV184-dS (lane 2), two distinct CSFV-specific signals were detected which corresponds to the anti-dS Western blot analysis (Fig. 3) and could be explained by the presence of N-glycans.

Western blot analysis of crude cell lysates probed with anti-CSFV E2 mAB. Samples originated from strains D#53 (lane 1), D#73 (lane 2) and D#79 (lane 3) co-producing the dS and the indicated fusion protein and A#299 (lane 4) producing the dS but no fusion protein. M, molecular weight marker
VLP formation
Formation of chimeric VLP was analyzed by ultracentrifugation. For each of the strains listed in Table 4, the dS and the fusion protein accumulated in the boundary layer of the two sucrose solutions during sucrose cushion ultracentrifugation. Additionally, the target proteins isolated from this boundary layer were detected in the same fractions after subsequent CsCl density gradient ultracentrifugation. They were co-separated from Hansenula HCP due to lower density (1.1–1.2 g cm−3). Thus, chimeric VLP formation of the dS and every fusion protein co-expressed was indicated. Co-localization and co-separation from HCP during the ultracentrifugation steps was also observed for the three heterologous proteins dS, FeLVp45-dS and FeLVp15E-dS co-expressed in strain M#4-5. This indicated formation of a three-component chimeric VLP.
Production of plain dS VLP using H. polymorpha
Hansenula polymorpha known as a potent host for recombinant HBsAg VLP production was shown to be well-suited for production of dS-based VLP. For demonstration and to set a benchmark for recombinant dS-based VLP production and purification, plain dS VLP were purified from strain A#299 in laboratory scale. Separation of the product from HCP contaminants in CsCl density gradient centrifugation is qualitatively shown in Fig. 5. This indicated the dS protein produced assembled into VLP structures characterized by a buoyant density of 1.14–1.17 g cm−3 which is consistent with a lipoprotein. Non-optimized fed batch fermentation yielded 35.2 ± 0.6 g L−1 dcw concentration during 70.5 h cultivation time. Using 70.4 ± 4 g dcw as biomass, 44.6 ± 2 mg VLP could be isolated. Analysis of the final preparation by SDS-PAGE followed by Coomassie staining or Western blotting, TEM imaging and DLS are shown in Fig. 6. Densitometric analysis of the Coomassie stained PAA gel (Fig. 6a, lane 3) indicated high purity (> 95%) which was confirmed by anti-HCP ELISA. This corresponds to a specific yield (YP/X) of 0.63 ± 0.07 mg g−1 of dS VLP per biomass. In the purified sample, the dS subunits of the VLP appeared primarily as monomeric proteins with an apparent MW of 14–15 kDa which is slightly below its theoretical MW (18.2 kDa). However, dS oligomers with higher apparent MW as well as dS-HMF with lower apparent MW than the monomer were detected in Coomassie stained PAA gel and dS-specific Western blot.

CsCl density gradient separation of HCP from plain dS VLP derived from recombinant H. polymorpha strain A#299 analyzed by Western blot. The ultracentrifugation tube was divided into 11 fractions with increasing density from fraction 1–11. Top: unspecific staining of proteins (Ponceau S). Bottom: anti-dS immunostaining using 7C12 mAB. M, molecular weight marker

Analysis of plain dS VLP purified from recombinant H. polymorpha strain A#299. a Western blot probed with anti-HCP serum (10 µg protein loaded) or anti-dS mAB 7C12 (1 µg protein loaded), respectively and Coomassie stained PAA gel analysis (10 µg protein loaded); M, molecular weight marker; HMF, higher mobility forms. b TEM images at 50,000-fold (top) and 250,000-fold (bottom) magnification. c DLS data after regularization analysis
The serum used in anti-HCP Western blot showed slight cross reactivity to the dS (Fig. 6a, lane 1). By loading 10 µg protein on the gel used for Western blotting, 14 individual HCP-specific signals could be detected by densitometry. DLS proved monomodal and monodisperse sample constitution (polydispersity index, PDI of 0.05) dominated by particles of 59 nm hydrodynamic diameter which was in good accordance with results from TEM imaging. Quantification of lipids yielded 0.79 ± 0.1 mg per mg protein which is equivalent to ~ 44% lipid content of the VLP.
Production of chimeric VLP
The formation of chimeric VLP development could be demonstrated for all viral antigens summarized in Table 4 by analytical ultracentrifugation. Exemplarily, chimeric VLP were purified at several mg scale either from strain T#3-3 expressing EDIIIWNV-dS or strain D#79 expressing E2CSFV102-dS, respectively. The DSP for chimeric VLP purification was simplified compared to plain dS VLP production. Desorption of the product from the Aerosil matrix was allowed at RT and no ion exchange chromatography was performed prior to preparative CsCl density gradient centrifugation.
Chimeric VLP with EDIIIWNV-dS
For the chimeric VLP originating from strain T#3-3, dialysis and size exclusion chromatography (SEC) were compared for desalting after CsCl density gradient separation. SEC proved to serve as polishing step and increased VLP purity on protein level by about 9% to > 95% compared to desalting by dialysis based on analysis by densitometry of Coomassie stained gels (Fig. 7c, lanes 12 and 13). The specific product yield (YP/X) was lowered by SEC by about 40% compared to desalting by dialysis from 0.7 to 0.4 mg chimeric VLP per g dcw. Based on the results obtained by densitometry, the chimeric VLP of both preparations consist of approximately 12% EDIIIWNV-dS and 88% wild-type dS. The chimeric VLP originating from SEC were characterized in more detail by TEM, DLS, CsCl density gradient separation and in dot blot assay as shown in Fig. 7. Analysis by TEM confirmed the formation of particles of predominantly 40–50 nm according to manual evaluation. Size distribution analysis by DLS detected a dominating particle population characterized by a hydrodynamic diameter of 67 nm (PDI 0.13) which corresponds well with the TEM imaging analysis. As expected for chimeric VLP, the dS and the fusion protein EDIIIWNV-dS accumulated in the same fractions during CsCl density gradient centrifugation with plausible buoyant densities for lipoproteins of 1.14–1.16 g cm−3.

Characterization of chimeric VLP isolated from strain T#3-3 co-producing dS and EDIIIWNV-dS and desalted by SEC. a TEM image after negative staining (100,000-fold magnification); b DLS data after regularization analysis; c lanes 1–11: Western blot analysis of fractions harvested from analytical CsCl density gradient separation (density increases gradually from lanes 1–11) probed with anti-dS mAB 7C12; Coomassie stained PAA gels for analysis of final VLP preparations after desalting by dialysis (lane 12) or SEC (lane 13), 10 µg protein loaded; d dot blot analysis of the native sample desalted by SEC and probed with anti-WNV mAB, position 1: chimeric VLP displaying the WNV antigen, position 2: plain dS VLP as a negative control
Additionally, the display of the WNV antigen on the VLPs’ surface was shown by dot blot analysis under native conditions.
Chimeric VLP with E2CSFV102-dS
Processing cell paste from strain D#79 yielded 0.5 g chimeric VLP per g dcw composed of approximately 33% E2CSFV102-dS and 67% wild-type dS according to analysis by densitometry of a Coomassie stained PAA gel (Fig. 8a, lane 12). Purity of the chimeric VLP preparation was ~ 75% based on the same analysis. The behavior of the VLP displaying the CSFV antigen in CsCl density gradient separation (Fig. 8a) was highly comparable to that of plain dS VLP (Fig. 5) and chimeric VLP displaying the WNV antigen (Fig. 7a): the two target proteins accumulated in the same fractions with buoyant densities of 1.14–1.16 g cm−3. Integrity of the VLP was demonstrated by TEM imaging detecting spherical particles of predominantly 50–70 nm. This was in good correlation with the size distribution analysis by DLS which verified monodisperse monomodal sample constitution characterized by a hydrodynamic diameter of 73 nm and a PDI of 0.11. Additionally, super-resolution imaging by N-SIM using specific antibodies demonstrated the accessibility of dS (Fig. 9a) and CSFV E2 (Fig. 9b) epitopes under native conditions. The signals from labeling each of dS and CSFV E2 co-localized in the same nano-scale structures (Fig. 9c) suggesting the display CSFV antigen on the particles’ surface. The SIM system was calibrated with 100 nm fluorescent beads (TetraSpeck™ microspheres, Thermo Fisher Scientific, Waltham, USA) which showed an apparent diameter of about 150 nm in the raw images (data not shown). The detected structures in Fig. 9 were either round shaped and of about 250 nm in diameter (representatively marked by white arrows) or larger (400–800 nm, marked by green arrows) and irregular shaped. Complexes of primary and secondary antibody were reported to stretch out for up to 30 nm [65] which may result in a larger apparent diameter of antibody decorated individual VLP.

Evaluation of chimeric VLP formation from material originated from strain D#79 co-producing dS and E2CSFV102-dS. a Lanes 1–11: Western blot analysis of fractions harvested from CsCl density gradient separation (density increases gradually from lanes 1–11), probed with anti-dS mAB 7C12; Lane 12: Coomassie stained PAA gel of pooled and desalted fractions 3 and 4. b TEM image after negative staining (100,000-fold magnification); c DLS data after regularization analysis

Chimeric VLP isolated from strain D#79 composed of dS and E2CSFV102-dS were analyzed under native conditions by N-SIM. Two series of images obtained from the same sample are presented showing fluorescence immunolabeling of dS in green (a-1, a-2), CSFV E2 antigen in red (b-1, b-2) and co-localization of the two labels in superimposed images in yellow (c-1, c-2). In each series of images two spots were consistently marked by arrows: signals of the size expected for individual VLP (white); largest signals in the respective frame (green)
Stability assessment of chimeric VLP
Shelf life and stability are generally critical for biotechnological products. Especially in the case of chimeric VLP incorporating large foreign antigens, VLP integrity over time or at elevated temperatures appears questionable. Both aspects were therefore investigated separately in our study (Fig. 10). Chimeric VLP isolated from strain D#79 and composed of dS and E2CSFV102-dS were analyzed by DLS and Western blot (Fig. 10a, b) immediately after preparation (“fresh”) and after 6 months of storage at 4–8 °C. Both methods manifested only minor changes of the samples’ constitution: A slight increase of hydrodynamic diameter (from 73 to 82 nm) as well as PDI (from 0.11 to 0.14) could be verified by DLS. Additionally, the fusion protein-specific signal in anti-dS and anti-CSFV E2 Western blot appeared more diffuse or as a pair of bands running closely to each other in the sample stored for 6 months compared to the freshly analyzed sample. However, loss of particulate character or substantial degradation of the product proteins was not observed.

Stability assessment of chimeric VLP. a, b DLS and Western blot analysis of real time stability experiment of purified VLP composed of dS and E2CSFV102-dS, isolated from strain D#79 and formulated at mg mL−1 protein concentration in desalting buffer. Data of fresh sample analysis are compared to data collected after 6 months of storage at 4–8 °C by independent but volume-normalized Western blot analysis. c DLS analysis of plain dS VLP and different chimeric VLP analyzed during step-wise increasing temperature allowing 5 min equilibration time in between the measurements
Thermal stability was tested by DLS for four different VLP preparations (Fig. 10c): plain dS VLP and chimeric VLP containing E2BVDV196-dS, E2CSFV102-dS or EDIIIWNV-dS. The determined hydrodynamic diameter of each of the different VLP changed only marginally during step-wise increase of the chamber temperature from 25 to 45 °C. For plain dS VLP and chimeric VLP containing E2CSFV102-dS or EDIIIWNV-dS, the diameter appears to slightly increase with increased temperature. This is most likely due to enhanced VLP collisions and therewith apparently reduced speed of Brownian motion. However, pronounced increase in hydrodynamic diameter indicating onset of VLP deformation or aggregation could be observed upon temperature increase from 45 to 50 °C for all VLP preparations. Complete aggregation occurred at 50 °C for chimeric VLP containing E2BVDV196-dS or EDIIIWNV-dS, at 55 °C for plain dS VLP or at 60 °C for chimeric VLP having the E2CSFV102-dS fusion protein incorporated, respectively.
Discussion
In this work, VLP formed by the dS were shown to be an effective platform for rational development of chimeric VLP displaying a variety of large foreign antigens. For the establishment of a robust platform, the methylotrophic yeast H. polymorpha proved to perform as a reliable microbial cell factory; none of the constructed fusion proteins failed to be co-expressed with the dS. During recombinant production, the fusion proteins were shown to be exposed to the lumen of the yeasts’ ER or Golgi system. They accumulated intracellularly and carried N-glycans if they had potential N-glycosylation sites within their amino acid sequence. Based on this, it can be assumed that the mechanism of dS-based VLP formation in recombinant H. polymorpha is highly comparable to the morphogenesis of HBsAg VLP in methylotrophic yeast Pichia pastoris [66]. The product proteins presumably accumulate in the yeasts’ subcellular membrane structures and congregate during DSP to plain dS VLP or chimeric VLP, respectively.
Chimeric VLP formation required co-production of the dS and a fusion protein in a single recombinant host. Therefore, a toolbox of strain generation strategies independent of antibiotic resistance genes was established. Isolation of heterologous H. polymorpha strains stably co-producing the heterologous proteins was allowed within only a single sequence of yeast transformation and subsequent strain selection allowing fast and simple generation of recombinant strains. Production levels of the respective fusion protein and the dS were observed to differ among the isolated yeast strains. Especially in the case of the fusion protein E2CSFV102-dS, the efficiency of chimeric VLP solubilization during cell disruption was found to be dependent on the relative expression levels of the fusion protein and the dS. The reason for reduced efficiency of target protein solubilization in case of higher relative amounts of the fusion protein remains ambiguous. It can be argued that the solubilization of dS and E2CSFV102-dS strongly depended on chimeric VLP formation since both proteins are membrane spanning and thus rather unlikely to be solubilized as monomers during cell disruption. It is believed that an excess of dS over the fusion protein is essential for chimeric dS-based VLP formation [13, 14] although detailed studies on this have not yet been published. Probably, incorporation of foreign antigens into the dS VLP scaffold is limited by steric issues which may arise if the density of foreign antigens within a VLP-forming structure exceeds a certain threshold. Interestingly, in these cases formation of dS VLP without or with low relative amounts of E2CSFV102-dS was not observed. This indicated that the two target proteins accumulated intracellularly in close proximity to each other and interacted with one another prior to cell lysis. However, the observed variety among the isolated and characterized production strains (Fig. 2), allowed us to pick the strain best suited for the integration into the DSP [31].
No protein purification tags were used during DSP which is highly desired for most applications especially for vaccines or pharmaceutical products [67]. Also, cost-intensive steps like immunoaffinity chromatography were not required here. Nevertheless, elimination of the costly CsCl density gradient purification step appears desirable to further improve cost efficiency.
Processing of cell paste from strain A#299 yielded plain dS VLP of similar quality (> 95% purity) and yield per biomass (0.63 ± 0.07 mg g−1) compared to literature on HBsAg VLP purification (~ 0.6 mg g−1, [68]). However, final recovery per culture volume was lower (22.3 ± 2 vs. ~ 50 mg L−1) due to non-optimized fermentation procedure applied in this study in contrast to carefully optimized fermentation protocol for HBsAg VLP production [69]. Since H. polymorpha is well known to be industrially applicable and to grow beyond 100 g dcw L−1 culture volume [70], improvements regarding the volume-normalized product yield can be expected after fermentation optimization. Additionally, the use of a synthetic growth medium during fermentation is highly desirable regarding regulatory approval for production of bio-pharmaceuticals [71]. The lipid content (~ 44%), the dimensions (59 nm hydrodynamic diameter) and the buoyant density (1.14–1.17 g cm−3) of Hansenula-derived dS VLP showed high similarity to what is described for natural occurring DHBV VLP (30–40%; 35–60 nm; 1.14–1.6 g cm−3) [20, 72].
Chimeric VLP presenting different foreign antigens could be purified by applying basically the same DSP that was used for purification of dS VLP before with similar product yields and protein purity. The purified chimeric VLP contained 33% E2CSFV102-dS or 12% EDIIIWNV-dS respectively, which is reasonable in the context of chimeric VLP vaccines [73]. We can only speculate about the number of VLP-forming protein subunits per individual VLP. The spherical ~ 22 nm HBV VLP contain approximately 100 HBsAg molecules [74]. Thus, dS-based VLP presumably contain well over 100 protein subunits due to their larger dimensions.
The foreign antigens of both chimeric VLP preparations containing either EDIIIWNV-dS or E2CSFV102-dS were shown to be accessible for immunolabeling under native conditions (Figs. 7d, 9). These assays suggest surface exposure of the foreign antigens on VLP. In addition, analysis by N-SIM demonstrated co-localization of the fusion protein and the dS in the same nano-scale particles. While the resolution may not be sufficient to localize both proteins in individual VLP, the authors conclude that co-localization in structures representing clusters of few VLP would support the presence of both proteins in individual VLP due to the physicochemical homogeneity of the analyzed sample.
Thermal stability of the recombinant VLP preparations was demonstrated (Fig. 10c) and could be explained by high similarity in their physicochemical properties compared to the native DHBV VLP. Since VLP are complex structures, multiple factors like mode of VLP purification, type and content of fusion protein and lipid content probably affect thermal stability which precludes simple explanation of the slight differences detected by this analysis (Fig. 10c). Preliminary 6 months real time stability data of chimeric VLP in simple PBS-like buffer support the use of this platform for vaccine development purposes. However, the potential application of the developed chimeric VLP as veterinary vaccine candidates cannot be shown without immunization and animal challenge studies. This represents the key task for the near future to extend the antigen presentation platform into a vaccine development platform.
Conclusions
This study describes the establishment of a robust and versatile VLP platform for presentation of large antigens. Based on the methylotrophic yeast H. polymorpha, it allows rational design, cost-effective production and purification of chimeric VLP. A variety of antigens originating from different animal-infecting viruses and described as highly immunogenic was successfully incorporated into a stable VLP scaffold formed by the dS. The obtained product yields make this technology a seriously competitive VLP development platform that should be considered for veterinary DIVA vaccine development in the future.
References
McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. Human hepatitis B vaccine from recombinant yeast. Nature. 1984;307:178–80.
Zhao Q, Li S, Yu H, Xia N, Modis Y. Virus-like particle-based human vaccines: quality assessment based on structural and functional properties. Trends Biotechnol. 2013;11:654–63.
Rohovie MJ, Nagasawa M, Swartz JR. Virus-like particles: next-generation nanoparticles for targeted therapeutic delivery. Bioeng Transl Med. 2016;2:43–57.
Yildiz I, Shukla S, Steinmetz NF. Applications of viral nanoparticles in medicine. Curr Opin Biotechnol. 2011;22:901–8.
Liu F, Ge S, Li L, Wi X, Liu Z, Wang Z. Virus-like particles: potential veterinary vaccine immunogens. Res Vet Sci. 2012;93:553–9.
Grgacic EVL, Anderson DA. Virus-like particles: passport to immune recognition. Methods. 2006;40:60–5.
Ulmer JB, Valley U, Rappuoli R. Vaccine manufacturing: challenges and solutions. Nat Biotechnol. 2006;24:1377–83.
Tan M, Jiang X. Subviral particle as vaccine and vaccine platform. Curr Opin Virol. 2014;6:24–33.
Morton DB. Vaccines and animal welfare. Rev Sci Techn. 2007;26:157–63.
European Medicines Agency. CVMP strategy on antimicrobials 2016–2020 (Reference number: EMA/CVMP/209189/2015). London: European Medicines Agency; 2016.
Meeusen ENT, Walker J, Peters A, Pastoret PP, Jungersen G. Current status of veterinary vaccines. Clin Microbiol Rev. 2007;20:489–510.
Rodríguez-Limas WA, Sekar K, Tyo KEJ. Virus-like particles: the future of microbial factories and cell-free systems as platforms for vaccine development. Curr Opin Biotechnol. 2013;24:1–5.
Anderson DA, Grgacic EVL. Viral vectors expressing fusion of viral large envelope protein and protein of interest. 2004. WO/2004/092387.
Grgacic EVL, Anderson DA, Loke P, Anders R. Recombinant proteins and virus-like particles comprising L and S polypeptides of avian hepadnaviridae and methods, nucleic acid constructs, vectors and host cells for producing same. 2006. WO/2008/025067A1.
Crisci E, Bárcena J, Montoya M. Virus-like particles: the new frontier of vaccines for animal viral infections. Vet Immunol Immunopathol. 2012;148:211–25.
Frietze KM, Peabody DS, Chackerian B. Engineering virus-like particles as vaccine platforms. Curr Opin Virol. 2016;18:44–9.
Grgacic EVL, Kuhn C, Schaller H. Hepadnavirus envelope topology: insertion of a loop region in the membrane and role of S in L protein translocation. J Virol. 2000;75:2455–8.
Mason WS, Seal G, Summers J. Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. J Virol. 1980;63:829–36.
Grgacic EVL, Anderson D. St, a truncated envelope protein derived from the S protein of duck hepatitis B virus, acts as a chaperone for the folding of the large envelope protein. J Virol. 2005;79:5346–52.
Schlicht HJ, Kuhn C, Guhr B, Mattaliano RJ, Schaller H. Biochemical and immunological characterization of the duck hepatitis B virus envelope proteins. J Virol. 1987;61:2280–5.
Mason WS, Gerlich WH, Taylor JM, Kann M, Mizokami T, Loeb D, Sureau C, Magnius L, Norder H. Hepadnaviridae. In: King AMQ, Adams AJ, Carstens EB, Lekowitz EJ, editors. Virus taxonomy. London: Elsevier; 2013. p. 445–55.
Yuasa S, Cheung RC, Pham Q, Robinson WS, Marion PL. Peptide mapping of neutralizing and nonneutralizing epitopes of duck hepatitis B virus pre-S polypeptide. Virology. 1991;181:14–21.
Jilbert AR, Kotlarski I. Immune responses to duck hepatitis B virus infection. Dev Comp Immunol. 2000;24:285–302.
Suh SO, Zhou JJ. Methylotrophic yeasts near Ogataea (Hansenula) polymorpha: a proposal of Ogataea angusta comb. nov. and Candida parapolymorpha sp. nov. FEMS Yeast Res. 2010;10:631–8.
Fuenmayor J, Gòdia F, Cervera L. Production of virus-like particles for vaccines. N Biotechnol. 2017;39:174–80.
Ahmad M, Hirz M, Pichler H, Schwab H. Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol. 2014;98:5301–17.
Roldão A, Mellado MCM, Castilho LR, Carrondo MJT, Alves PM. Virus-like particles in vaccine development. Expert Rev Vaccines. 2010;9:1149–76.
Liu F, Wu X, Li L, Liu Z, Wang Z. Use of baculovirus expression system for generation of virus-like particles: successes and challenges. Protein Expr Purif. 2013;2013(90):106–16.
Heijtink RA, van Bergen P, Melber K, Janowicz ZA, Osterhaus ADME. Hepatitis B surface antigen (HBsAg) derived from yeast cells (Hansenula polymorpha) used to establish an influence of antigenic subtype (adw 2, adr, ayw 3) in measuring the immune response after vaccination. Vaccine. 2002;20:2191–6.
Janowicz ZA, Melber K, Merckelbach A, Jacobs E, Harford N, Comberbach M, Hollenberg CP. Simultaneous expression of the S and L surface antigens of hepatitis B, and formation of mixed articles in the methylotrophic yeast, Hansenula polymorpha. Yeast. 1991;7:431–43.
Schaefer S, Piontek M, Ahn SJ, Papendieck A, Janowicz ZA, Timmermans I, Gellissen G. Recombinant hepatitis B vaccines—disease characterization and vaccine production. In: Gellissen G, editor. Hansenula polymorpha—biology and applications. Weinheim: Wiley-VCH; 2002. p. 175–210.
Mignon C, Sodoyer R, Werle B. Antibiotic-free selection in biotherapeutics: now and forever. Pathogens. 2015;4:157–81.
Lindberg A, Brownlie J, Gunn GJ, Houe H, Moennig V, Saatkamp HW, Sandvik T, Valle PS. The control of bovine viral diarrhoea virus in Europe: today and in the future. Rev Sci Tech Off Int Epiz. 2006;25:961–79.
Edwards S, Fukusho A, Lefèvre PC, Lipowski A, Pejsak Z, Roehe P, Westergaard J. Classical swine fever: the global situation. Vet Microbiol. 2000;73:103–19.
Reinacher M. Diseases associated with spontaneous feline leukemia virus (FeLV) infection in cats. Vet Immunol Immunopathol. 1989;21:85–95.
Kramer DL, Styer LM, Ebel GD. A global perspective on the epidemiology of west nile virus. Annu Rev Entomol. 2008;53:61–81.
Murray NEA, Quam MB, Wilder-Smith A. Epidemiology of dengue: past, present and future prospects. Clin Epidemiol. 2013;5:299–309.
Thrane S, Janitzek CM, Matondo S, Resende M, Gustavsson T, de Jongh WA, Clemmensen S, Roeffen W, van de Vegte-Bolmer M, van Gemert GJ, Sauerwein R, Schiller JT, Nielsen MA, Theander TG, Salanti A, Sander AF. Bacterial superglue enables easy development of efficient virus-like particle based vaccines. J Nanobiotechnol. 2016;14:1–6.
Smith MT, Hawes AK, Bundy BC. Reengineering viruses and virus-like particles through chemical functionalization strategies. Curr Opin Biotechnol. 2013;24:620–6.
Kang HA, Gellissen G. Hansenula polymorpha. In: Gellissen G, editor. Production of recombinant proteins. Weinheim: Wiley-VHC; 2005. p. 134.
Dagert M, Ehrlich SD. Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene. 1979;6:23–8.
Miller JH. Experiments in molecular genetics. 3rd ed. New York: Cold Spring Harbor Laboratory; 1972.
Lahtchev KL, Semenova VD, Tolstorukov II, van der Klei I, Veenhuis M. Isolation and properties of genetically defined strains of the methylotrophic yeast Hansenula polymorpha CBS4732. Arch Microbiol. 2002;177:150–8.
Morais JDF, Maia MHD. Estudos de microorganismos encocentrados em leitos de despejos de caldas de destilarias de Pernambuco. II. Uma nova especie de Hansenula, H. polymorpha. An Esc Super Quim Univ Recife. 1959;1:15–20.
Faber KN, Haima P, Harder W, Veenhuis M, Ab G. Highly-efficient electrotransformation of the yeast Hansenula polymorpha. Curr Genet. 1994;25:305–10.
Guengerich L, Kang HA, Behle B, Gellissen G, Suckow M. A platform for heterologous gene expression based on the methylotrophic yeast Hansenula polymorpha. In: Kueck U, editor. Genetics and Biotechnology. Berlin: Springer; 2004. p. 273–87.
Klingmüller U, Schaller H. Hepadnavirus infection requires interaction between the viral pre-S domain and a specific hepatocellular receptor. J Virol. 1993;67:7414–22.
Peterson GL. A Simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem. 1977;83:346–56.
Frings CS, Fendley TW, Dunn RT, Queen CA. Improved determination of total serum lipids by the sulfo-phospho-vanillin reaction. Clin Chem. 1972;18:673–4.
Pink M, Verma N, Rettenmeier AW, Schmitz-Spanke S. CBB staining protocol with higher sensitivity and mass spectrometric compatibility. Electrophoresis. 2010;31:593–8.
Bjerrum OJ, Schafer-Nielsen C. Buffer systems and transfer parameters for semidry electroblotting with a horizontal apparatus. In: Dunn MJ, editor. Electrophoresis. Weinheim: Verlag Chemie; 1986. p. 315–27.
Bolin SR. Immunogens of bovine viral diarrhea virus. Vet Microbiol. 1993;37:263–71.
Donis RO, Corapi W, Dubovi EJ. Neutralizing monoclonal antibodies to bovine viral diarrhoea virus bind to the 56 K to 58 K glycoprotein. J Gen Virol. 1987;69:77–86.
Hulst MM, Westra DF, Wensvoort G, Moormann RJ. Glycoprotein E1 of hog cholera virus expressed in insect cells protects swine from hog cholera. J Virol. 1993;67:5435–42.
van Zijl M, Wensvoort G, de Kluyver E, Hulst M, can der Gulden H, Gielkens A, Berns A, Moormann R. Live attenuated pseudorabies virus expressing envelope glycoprotein E1 of hog cholera virus protects swine against both pseudorabies and hog cholera. J Virol. 1991;5:2761–5.
Huang YL, Deng MC, Wang FI, Huang CC, Chang CY. The challenges of classical swine fever control: modifed live and E2 subunit vaccines. Virus Res. 2014;2014(179):1–11.
Li Y, Wang J, Kanai R, Modis Y. Crystal structure of glycoprotein E2 from bovine viral diarrhea virus. Proc Natl Acad Sci USA. 2013;110:6805–10.
Hossain MM, Rowland RR. Replicon particle expressing the E2 glycoprotein of bovine viral diarrhea virus immunization and evaluation of antibody response. Viral Immunol. 2017. https://doi.org/10.1089/vim.2017.0053.
Kikuchi M, Yoshimura K, Nakahama K. Production of human lysozyme. 1986. US4945051A.
Gavrilov BK, Rogers K, Fernandez-Sainz IJ, Holinka LG, Borca MV, Risatti GR. Effects of glycosylation on antigenicity and immunogenicity of classical swine fever virus envelope proteins. Virology. 2011;420:135–45.
Langhammer S, Hübner J, Kurth R, Denner J. Antibodies neutralizing feline leukaemia virus (FeLV) in cats immunized with the transmembrane envelope protein p15E. Immunology. 2006;117:229–37.
Langhammer S, Hübner J, Oswald J, Kurth R, Denner J. Immunization with the transmembrane protein of a retrovirus, deline leukemia virus: absence of antigenemia following challenge. Antivir Res. 2011;89:119–23.
Langhammer S, Fiebig U, Kurth R, Denner J. Increased neutralizing antibody response after simultaneous immunization with Leucogen and the feline leukemia virus transmembrane protein. Intervirology. 2011;54:78–86.
Chu JHJ, Chiang CCD, Ng ML. Immunization of Flavivirus west nile recombinant domain III protein induced specific immune response and protection against west nile virus infection. J Immunol. 2007;178:2699–705.
Loukanov AR, Emin S. Quantum dots for detection, identification and tracking of single biomolecules in tissue and cells. In: Tiwari A, Mishra AK, Kobayashi H, Turner APF, editors. Intelligent nanomaterials: processes, properties, and applications. Hoboken: Wiley; 2012. p. 671.
Lünsdorf H, Gurramkonda C, Adnan A, Khanna N, Rinas U. Virus-like particle production with yeast: ultrastructural and immunocytochemical insights into Pichia pastoris producing high levels of the hepatitis B surface antigen. Microb Cell Fact. 2011;10:1–16.
Arnau J, Lauritzen C, Petersen GE, Pedersen J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr Purif. 2006;48:1–13.
Gurramkonda C, Zahid M, Nemani SK, Adnan A, Gudi SK, Khanna N, Ebensen T, Lünsdorf H, Guzmán CA, Rinas U. Purification of hepatitis B surface antigen virus-like particles from recombinant Pichia pastoris and in vivo analysis of their immunogenic properties. J Chromatogr B. 2013;940:104–11.
Gurramkonda C, Adnan A, Gäbel T, Lünsdorf H, Ross A, Nemani SK, Swaminathan S, Khanna N, Rinas U. Simple high-cell density fed-batch technique for high-level recombinant protein production with Pichia pastoris: application to intracellular production of hepatitis B surface antigen. Microb Cell Fact. 2009;8:13.
Mayer AF, Hellmuth K, Schlieker H, Lopez-Ulibarri R, Oertel S, Dahlems U, van Strasser AW, Loon AP. An expression system matures: a highly efficient and cost-effective process for phytase production by recombinant strains of Hansenula polymorpha. Biotechnol Bioeng. 1999;63:373–81.
Hahn-Hägerdal B, Karhumaa K, Larsson CU, Gorwa-Grauslund M, Görgens J, van Zyl WH. Role of cultivation media in the development of yeast strains for large scale industrial use. Microb Cell Fact. 2005;4:31.
Cova L, Abdul F, Buronfosse T. Avihepadnavirus. In: Tidona C, Darai G, editors. The Springer index of viruses. Berlin: Springer; 2011. p. 615–24.
Gordon DM, McGovern TW, Krzych U, Cohen JC, Schneider I, LaChance R, Heppner DG, Yuan G, Hollingdale M, Slaoui M, Hauser P, Voet P, Sadoff JC, Ballou WR. Safety, immunogenicity, and efficacy of a recombinantly produced Plasmodium falciparum circumsporozoite protein-hepatitis B surface antigen subunit vaccine. J Infect Dis. 1995;171:1576–85.
Mangold CMT, Streeck RE. Mutational analysis of the cysteine residues in the hepatitis B virus small envelope protein. J Virol. 1993;67:4588–97.
Pugh JC, Di Q, Mason WS, Simmons H. Susceptibility to duck hepatitis B virus infection is associated with the presence of cell surface receptor sites that efficiently bind viral particles. J Virol. 1995;69:4814–22.
Gupta R, Jung E, Brunak S. Prediction of N-glycosylation sites in human proteins. 2004. http://www.cbs.dtu.dk/services/NetNGlyc/. Accessed 14 Oct 2017.
Schlecht-Louf G, Mangeney M, El-Garch H, Lacombe V, Poulet H, Heidmann T. A targeted mutation within the feline leukemia virus (FeLV) envelope protein immunosuppressive domain to improve a canarypox virus-vectored FeLV vaccine. J Virol. 2013;88:92–1001.
Authors’ contributions
DW, TR, MS, AK, MW, JL, CP and BK designed the fusion proteins and the experiments. DW, TR and JL performed most of the experiments and analyzed the data together with MP, VJ, MS and MW. CP, BK, JAC and JGB contributed the N-SIM analysis; AB contributed the TEM imaging. DW wrote the manuscript. MS, MP, JM, VJ, JL, JAC, and MW critically reviewed the manuscript. MP, JM, GS, MW, and VJ conceived the study and supervised the research. All authors read and approved the final manuscript.
Acknowledgements
The authors gratefully acknowledge Sylvia Denter, Margit Kombüchen, Aileen Krüger, Lisa Siebel, Michaela Steffenhagen and Elisabeth Wesbuer for technical assistance.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
The project was founded in the framework of ERA-Net EuroTransBio-8 (European Research Area Network, Code: 031A505B).
The Burnet Institute was supported by funding from the National health and medical Research Council of Australia (Program Grant and Fellowship to J. Beeson, and Independent Research Institutes Infrastructure Support Scheme) and a Victorian Government Operational Infrastructure Support grant.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wetzel, D., Rolf, T., Suckow, M. et al. Establishment of a yeast-based VLP platform for antigen presentation. Microb Cell Fact 17, 17 (2018). https://doi.org/10.1186/s12934-018-0868-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-018-0868-0